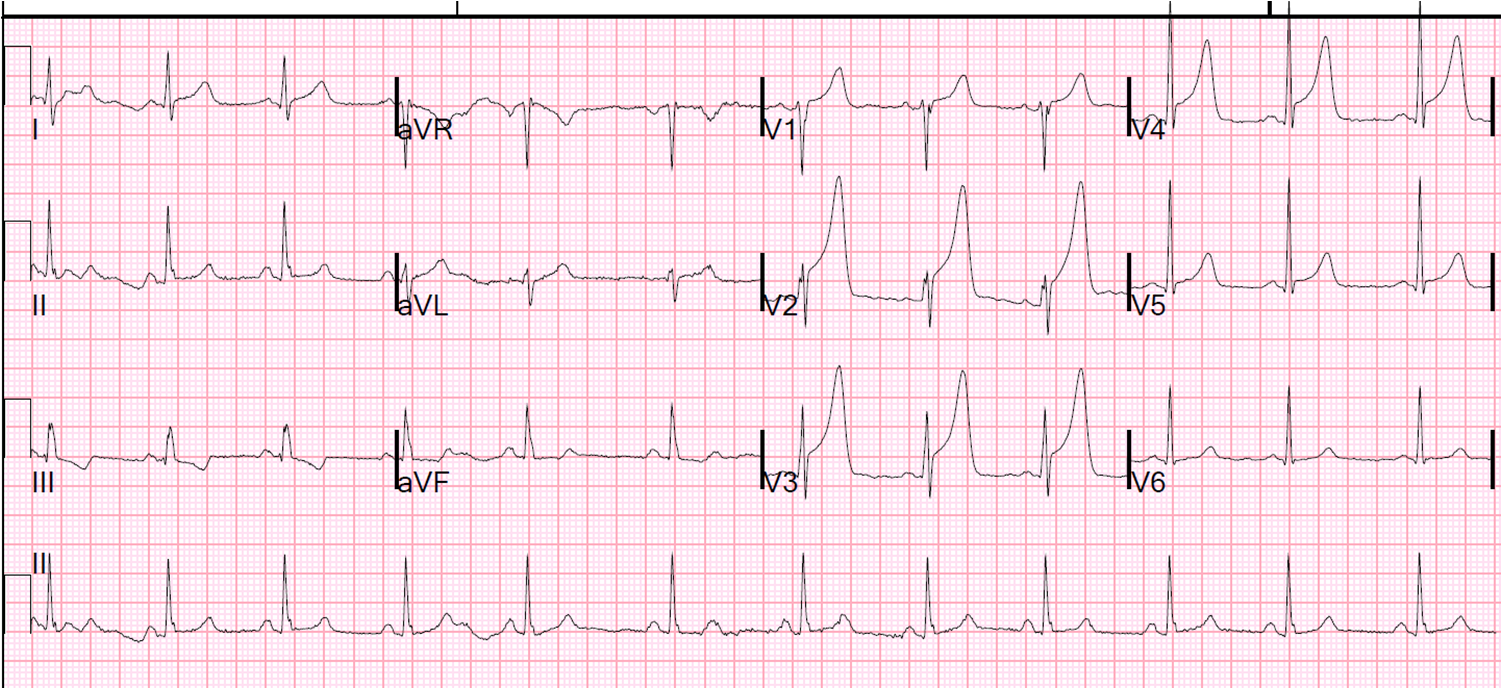
A male is his 30's with h/o HTN presented after 6 hours of chest pain. The pain was crushing and substernal, associated with nausea, vomiting, and diaphoresis, radiating to his back, 10 out of 10 in intensity, the "worst pains ever experienced". He described the sensation as "an elephant sitting on chest". Here is his EKG by EMS:
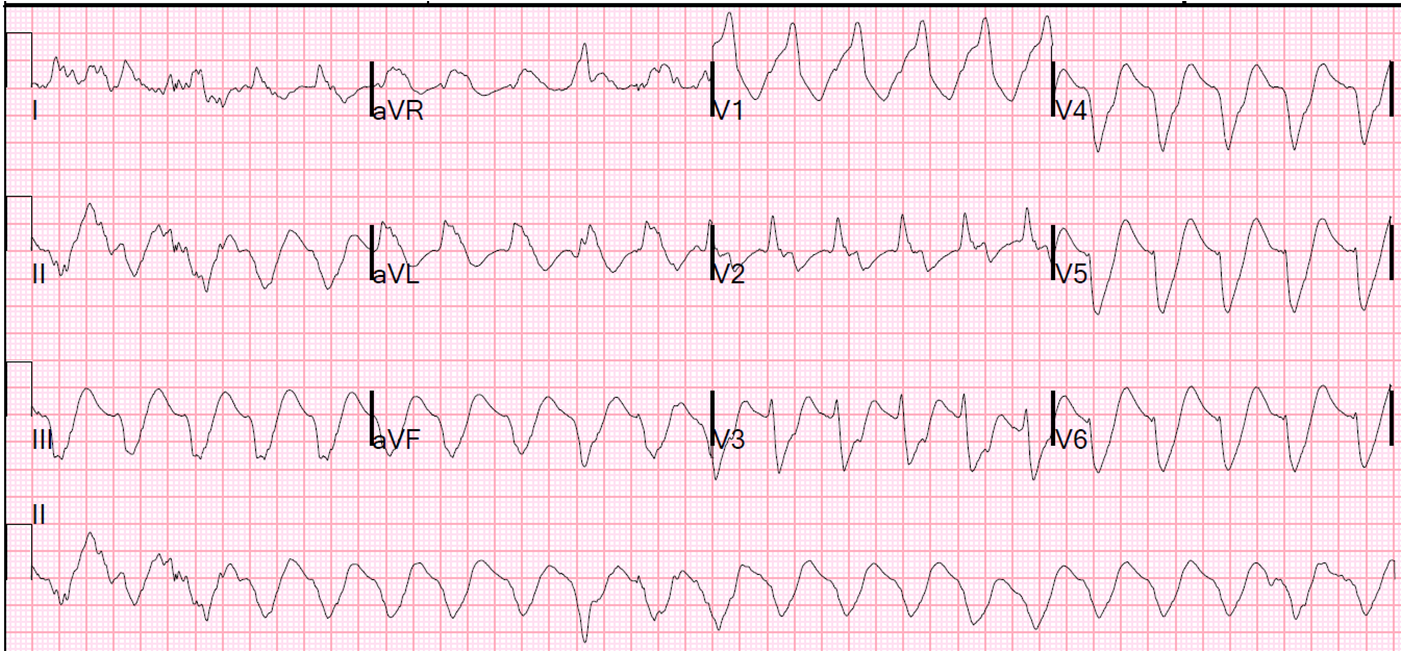
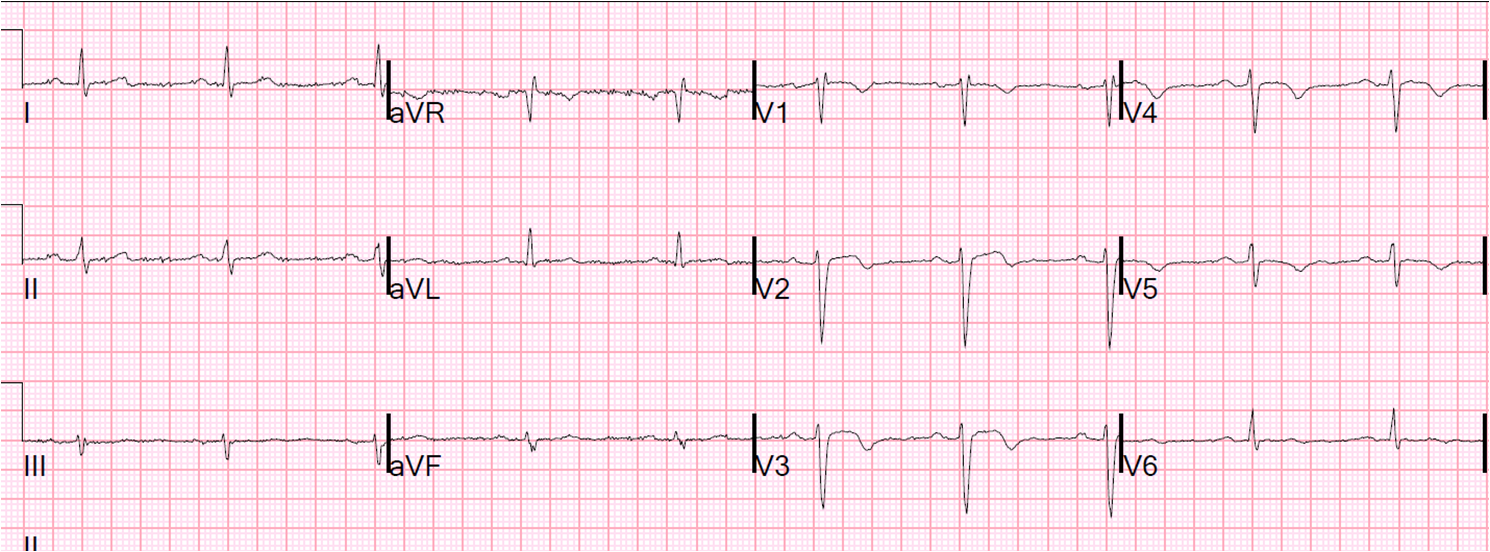
![]() |
| Obvious anterior STEMI with very large T-waves. Is this consistent with 5 hours of injury? |
The cath lab was activated prehospital. Patient received 324 aspirin and 3 nitroglycerin tablets sublingually prior to arrival to the ED with mild improvement of pain.
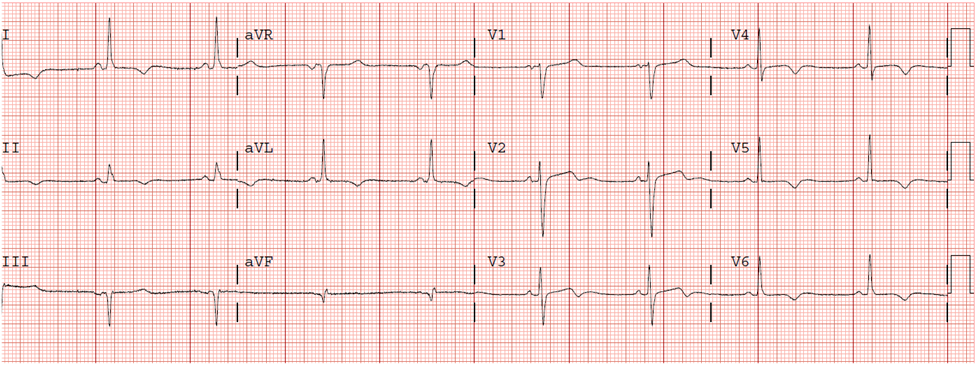
Here is the ED ECG:
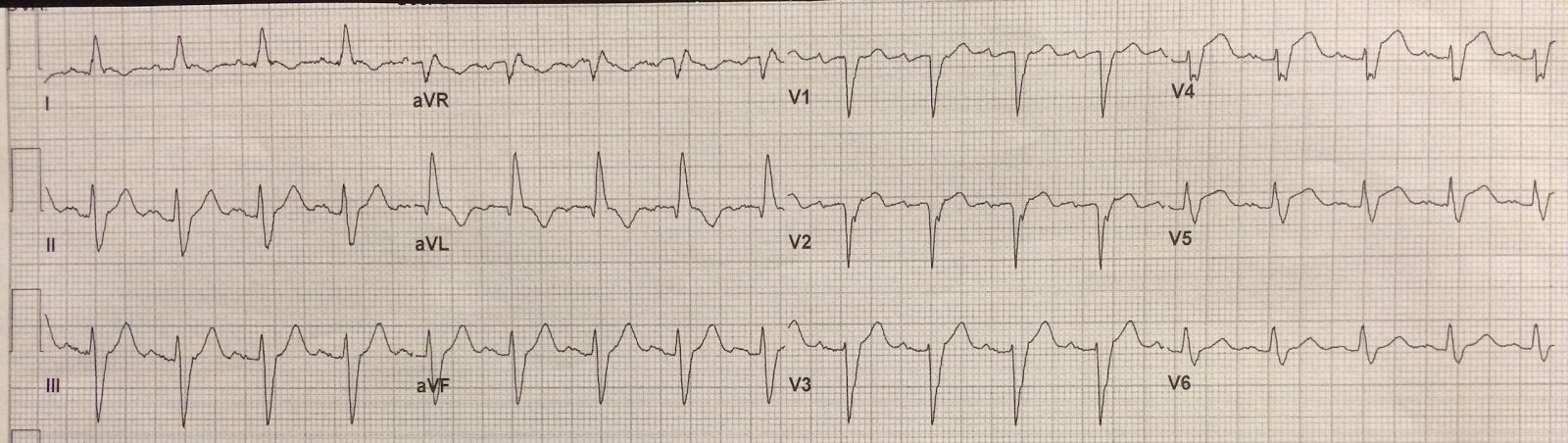
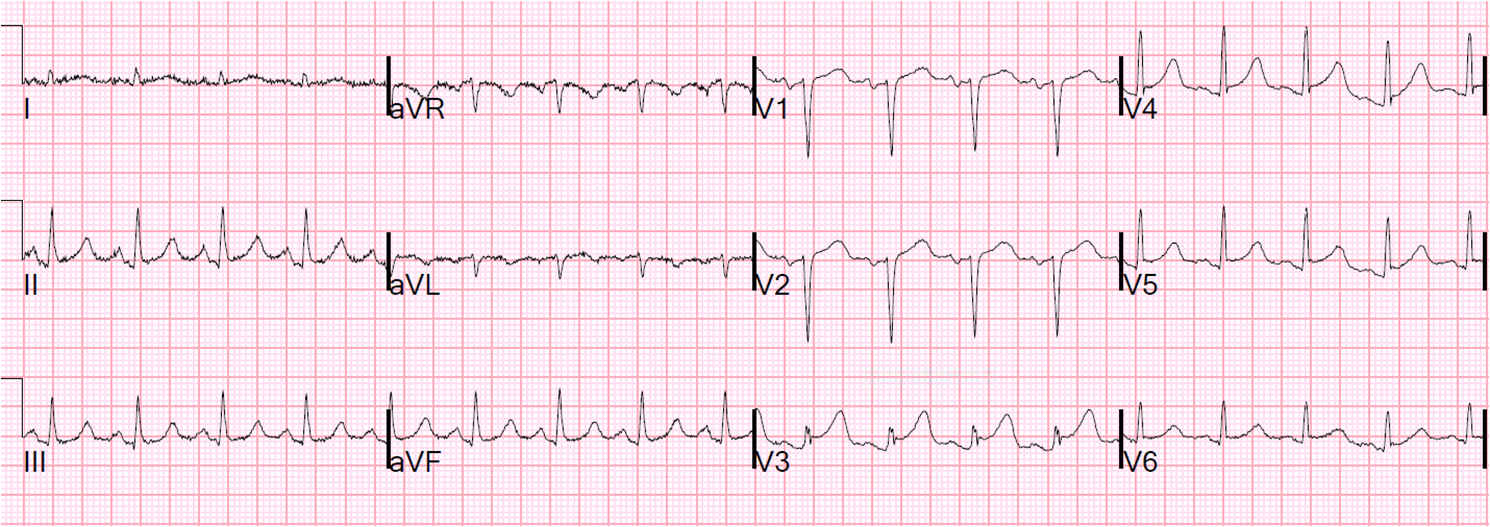
![]() |
| Very hyperacute T-waves and ST elevation. Somewhat diminished after Nitroglycerine. |
Suppose you were at a small hospital that only has thrombolytics. You have the option of giving thrombolytics or of transferring to a PCI capable hospital. The Door (small hospital) to Balloon (PCI hospital) time will be 90 minutes.
Should you give thrombolytics?
It is well known that thrombolytics work best with fresh thrombus, within 2 hours of onset of occlusion (Steg et al., see reference 8 below). But this patient has had 5 hours of chest pain.
However, the ECG shows a
very high level of "Acuteness," as demonstrated by 1) the size of the T-wave 2) the amount of ST elevation and 3) the absence of Q-waves. Hyperacute T-waves are present when there is a large amount of viable, salvageable, myocardium. As the myocardium infarcts (Q-waves), rather than being "injured," (as manifested by ST elevation and hyperacute T-waves), the size of the T-wave diminishes, and also the ST elevation diminishes and Q-waves begin to form (although in anterior STEMI Q-waves may form very early due to ischemia of the conducting system, not infarct!).
The most specific sign of a very early MI are hyperacute T-waves.We know that ACS is a very dynamic process, with opening and closing of the vessel. The time of onset of chest pain is not a good determinant of the time of onset of irreversible ischemia (infarct). We also know that pain is not a very reliable indicator of reperfusion/reocclusion. Studies of patients on 12-lead ST segment monitors shows rise and fall of ST segments while patients are asymptomatic.
So, this ECG is very hyperacute. Almost all the myocardium at risk is viable and can be salvaged. The thrombus has been forming and lysing spontaneously, which means that it is fresh thrombus, and very amenable to lysing with tPA or TNK-tPA.
If I saw this patient, assuming there are no contraindications, I would give thrombolytics (fibrinolytics) and transfer.
When the patient arrives at the PCI-capabel hospital, if there is no reperfusion, as measured by less than 50% ST-segment resolution in the single lead with maximum elevation, or clinical evidence of failed reperfusion within 90 minutes after fibrinolysis, then rescue coronary intervention can be performed.
See this article in the New England Journal.OutcomeThe patient went for angiogram/PCI, and a 100% mid-LAD occlusion was found and opened.
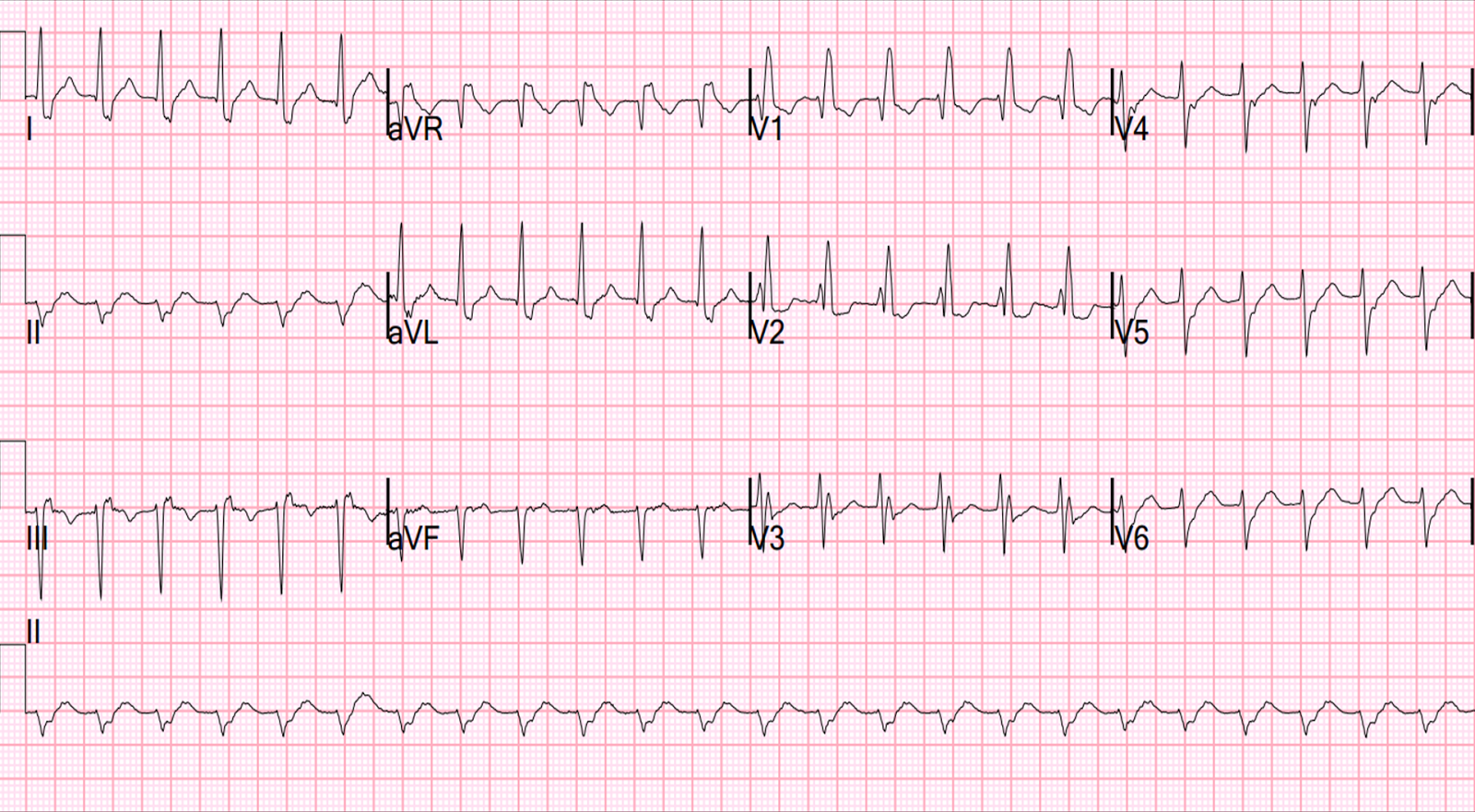
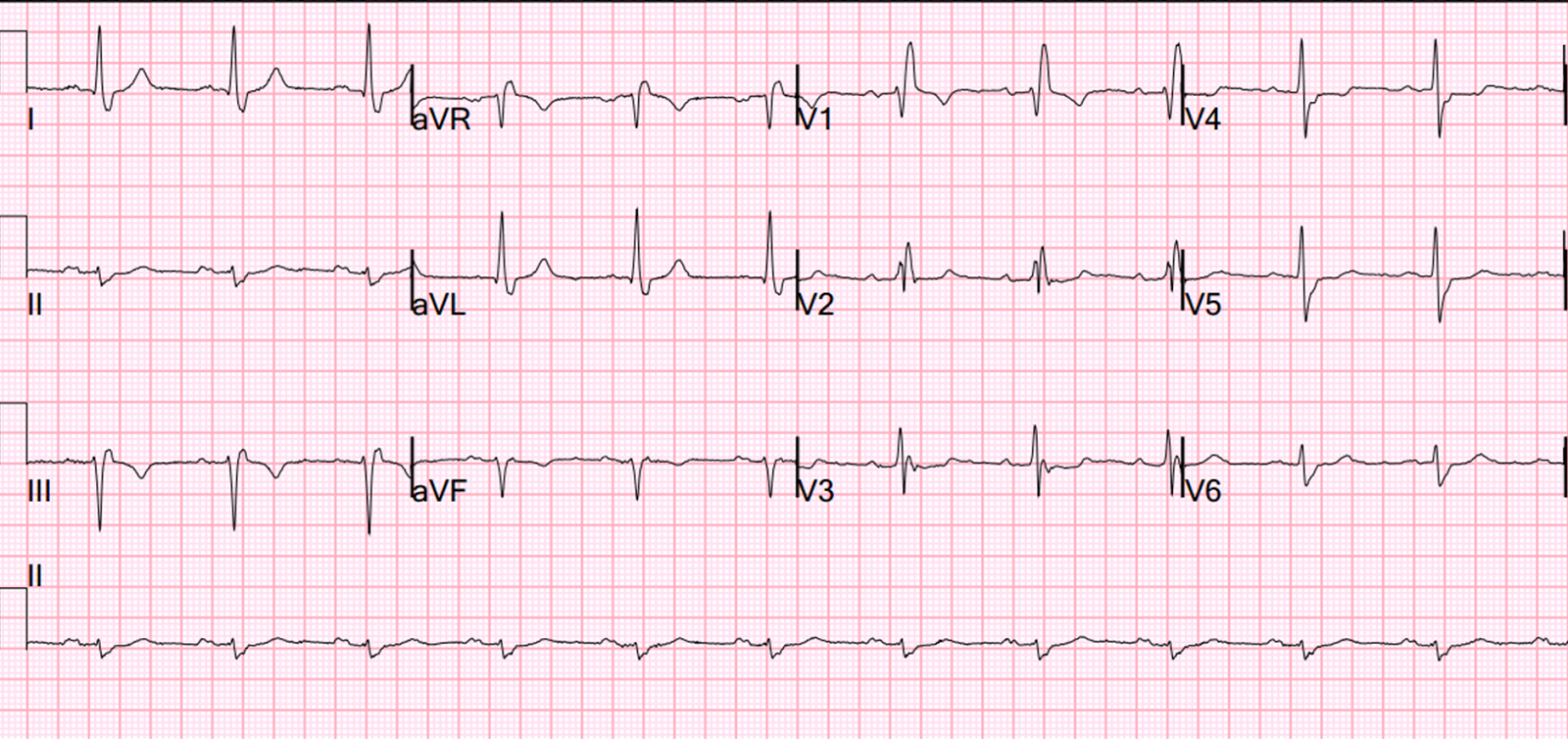
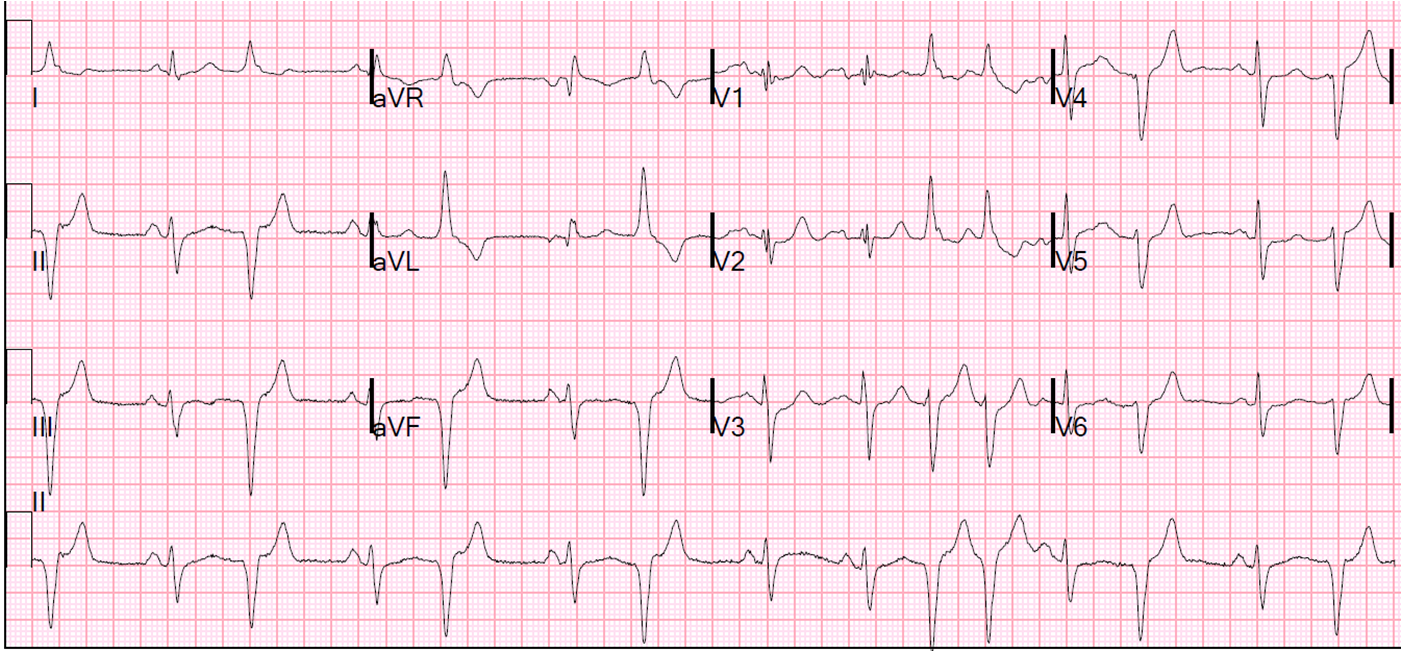
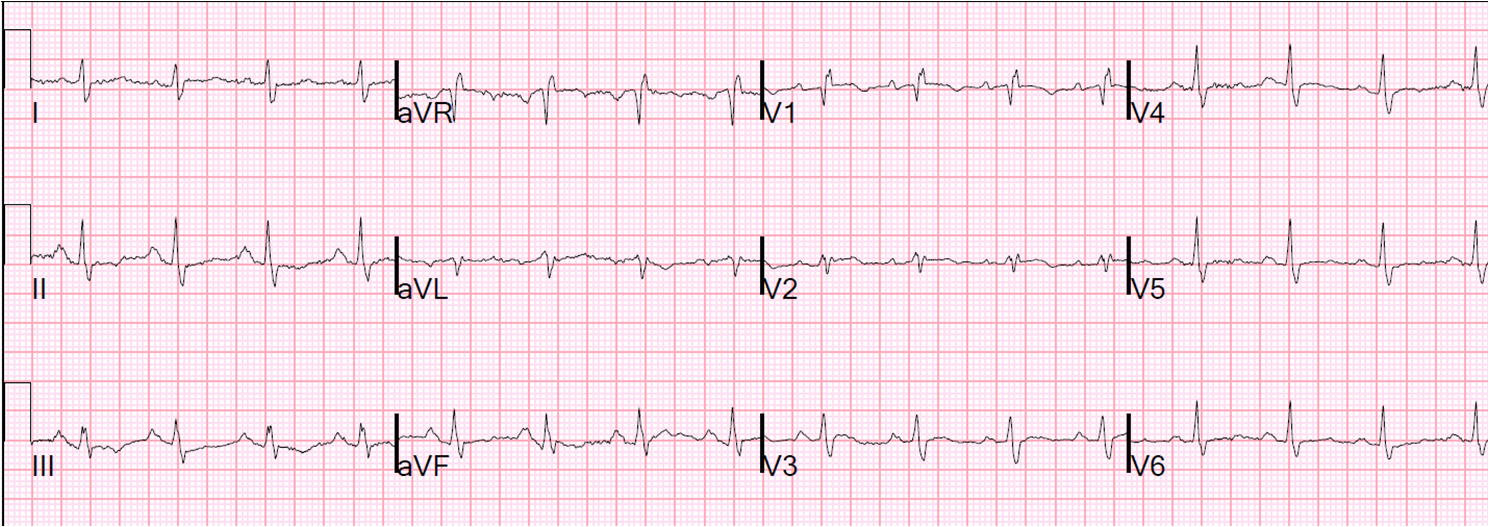
The peak troponin I was 0.9 ng/mL (very low). The ECG the next day is here:
![]() |
| There is subtle Wellens' waves in V2 and V3. These waves are signs of infarction. (It is my theory that Wellens' is simply spontaneously reperfused anterior STEMI in which the ST elevation is never recorded. This demonstrates this phenomenon well). |
Echocardiogram next day: There was a mild antero-apical wall motion abnormality. The convalescent echo, which is done after recovery of "stunned" myocardium at 4-6 weeks, will almost certainly be normal.
Of the significant myocardium at risk in the anterior wall, almost all of was salvaged because of the hyperacute nature of the injury, as manifested by huge T-waves, and the rapid reperfusion.
Lesson:1. The ECG is a better indicator of acuteness than the time since onset of chest pain.
2. When the ECG is very acute, thrombolytics are beneficial, and waiting for PCI has much less benefit, and probably relative harm.
Relevant references, in addition to the New England Journal article above
1. Engblom H, Strauss DG, Heden B, et al. The evaluation of an electrocardiographic myocardial ischemia acuteness score to predict the amount of myocardial salvage achieved by early percutaneous coronary intervention Clinical validation with myocardial perfusion single photon emission computed tomography and cardiac magnetic resonance. J Electrocardiol 2011;44(5):525-32.
2. Armstrong PW, Fu Y, Westerhout CM, et al. Baseline Q-wave surpasses time from symptom onset as a prognostic marker in ST-segment elevation myocardial infarction patients treated with primary percutaneous coronary intervention. J Am Coll Cardiol 2009;53(17):1503-9.
3. Sejersten M, Ripa RS, Maynard C, et al. Timing of ischemic onset estimated from the electrocardiogram is better than historical timing for predicting outcome after reperfusion therapy for acute anterior myocardial infarction: a DANish trial in Acute Myocardial Infarction 2 (DANAMI-2) substudy. Am Heart J 2007;154(1):61 e1-8.
4. Engblom H, Heden B, Hedstrom E, Wagner G, Arheden H. ECG Estimate Of Ischemic Acuteness and Time from Pain Onset for Predicting Myocardial Salvage in Patients Undergoing Primary Percutaneous Coronary Intervention. AHA Abstract 2404. Circulation 2007;116(Suppl II):II_528.
5. Wong CK, Gao W, Raffel OC, et al. Initial Q waves accompanying ST-segment elevation at presentation of acute myocardial infarction and 30-day mortality in patients given streptokinase therapy: an analysis from HERO-2. Lancet 2006;367:2061-7.
6. Gersh BJ, Stone GW, White HD, Holmes DR, Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005;293(8):979-86.
7. Taher T, et al. Aborted myocardial infarction in patient with ST segment elevation: Insights from the Assessment of the Safety and Efficacy of a New Thrombolytic Regimen-3 Trial Electrocardiographic Substudy. J Am Coll Card 2004;44:38-43.
8. Steg PG, Bonnefoy E, Chabaud S, et al. Impact of time to treatment on mortality after prehospital fibrinolysis or primary angioplasty: data from the CAPTIM randomized clinical trial. Circulation 2003;108(23):2851-6.
9. Heden B, Ripa R, Persson E, et al. A modified Anderson-Wilkins electrocardiographic acuteness score for anterior or inferior myocardial infarction. Am Heart J 2003;146(5):797-803.
10. Corey KE, Maynard C, Pahlm O, et al. Combined historical and electrocardiographic timing of acute anterior and inferior myocardial infarcts for prediction of reperfusion achievable size limitation. Am J Cardiol 1999;83(6):826-31.
11. Hochrein J, Sun F, Pieper KS, et al. Higher T-wave amplitude associated with better prognosis in patients receiving thrombolytic therapy for acute myocardial infarction (a GUSTO-1 substudy). Global Utilization of Streptokinase and Tissue plasminogen activator for Occluded Coronary Arteries. Am J Cardiol 1998;81(9):1078-84.
12. Wilkins ML, Pryor AD, Maynard C, et al. An electrocardiographic acuteness score for quantifying the timing of a myocardial infarction to guide decisions regarding reperfusion therapy. Am J Cardiol 1995;75(8):617-20.
13. Anderson ST, Wilkins M, Weaver WD, Selvester RH, Wagner GS. Electrocardiographic phasing of acute myocardial infarction. J Electrocardiol 1992;25 Suppl:3-5.